Many advancements have occurred in the field of gene therapy since the last webinar on this topic. The Eye & Ear Foundation’s June 25th webinar, “Seeing the Future of Gene Therapy,” focused on current options for clinical patients and new therapies.
Michelle Alabek, MS, LCGC, certified genetic counselor at the UPMC Vision Institute, began the presentation. Her clinical focus is inherited retinal dystrophies (IRDs). In addition to providing clinical genetic counseling, she coordinates the Retinal Dystrophy Clinic and operationally supports delivery of commercial and clinical trial gene therapies for IRDs.
Gene Therapy: The Patient Journey
“Why is gene therapy such a challenge?” Alabek asked. Getting the therapy to patients means getting the right gene to the right place at the right level without unwanted outcomes. This is not easy, especially with hoops to go through, like host barriers, the clinical trial process, vector selection, and manufacturing/demand.
Once there is a therapy, the first step is to identify eligible patients who might be interested. Each therapy has its own criteria. Common inclusion criteria for IRD gene therapy include gender, age, diagnosis, visual acuity, genetic result, and ocular testing results (i.e. visual field, ERG, OCT). There are also exclusion criteria; common ones include systemic disease, medications, prior eye surgeries, pregnant/breastfeeding, other eye conditions, other genetic diagnosis, and prior treatment with an investigational drug.
Even if patients meet all the inclusion criteria, if they meet any of the exclusion criteria, the treatment might not be a good option. A lot of testing is done on patients in the clinic every time they come in. This way, the data is on file to help the team assess potential eligibility as new therapies may become available.
The process for identifying eligible patients starts with the clinical team, which does the following:
- Review eligibility criteria
- Run potential patient report
- Assess available/missing data
- Arrange consult visit
- Perform updated/missing evaluations
- Eligibility determination
Patients are continuously updated and educated throughout the process. The team maintains a database of all of their patients that are seen in clinic so they can easily run reports. Ultimately, the patient decides if they want to move forward.
Coordinating Treatment
Once a patient is identified as potentially eligible, the next step is coordinating treatment. Naturally, this is also complex, because it involves many stakeholders (patients, consumers, clinical, manufacturer, financial, and operational). It takes all five of these groups of individuals working together and being on the same page to successfully treat a patient, Alabek said.
There are also hospital-wide implications, such as infrastructure, volume/capacity, and financial considerations. “We’re fortunate that at the new Vision Institute, there was a lot of forethought put into ensuring that we can deliver these types of therapies in a very efficient way with involvement and coordination,” Alabek said.
Access is also important; it includes patient awareness, cost of treatment, and the number and location of treatment centers. Patients who do not have a specialty center might not realize there are therapies for them. While treatment is often covered by insurance, “but when we talk about equity and fairness in the US and globally, access to these gene therapies is a very important topic that we need to make sure we’re paying attention to,” Alabek said. Additionally, there may only be a handful of centers in the U.S. participating in a trial or approved to deliver an FDA-approved gene therapy.
Process After Determining Eligibility
Financial Approval
- Benefits investigation
- In vs out-of-network
- Authorizations
- Contracts
Pre-treatment
- Baseline clinical assessments (often can be quite lengthy but very important)
- Informed consent
- Procure drug
Treatment (variables)
- Out-patient vs in-patient
- OR vs clinic
- Anesthesia
- Duration
- Medications
Post-treatment
- Clinical follow-ups
- Vision rehabilitation

As always, patients are continuously updated and educated throughout this process. When it comes to treatment, the schedule really starts once all the financial approvals are in place. It involves a lot of visits, but they are clinically necessary.
Role of Genetic Counselors
Alabek defined genetic counseling as the process of helping people understand and adapt to the medical, psychological, and familial implications of genetic contributions to disease. It involves ongoing support, a pre-test visit, coverage and testing, interpreting results, and a post-test visit. “I think we’re learning as a profession that that really is kind of translating into this gene therapy space and helping patients to navigate that process,” Alabek added.
Topics for Eligible Patients to Consider
- Anticipated outcomes
- Potential risks
- Availability does not equal eligibility
- Schedule/parameters of treatment
- Cost/coverage
- Uncertainty of long-term risks
- Other available options (are there other therapies coming down the pipeline for this condition? How do they compare to each other?)
- Emotional and physical toll
- Available support system
Gene Therapy for Retinal Degenerations: Promises and Challenges
The next part of the presentation was by Dr. José-Alain Sahel, MD, Chair and Distinguished Professor of the Department of Ophthalmology at the University of Pittsburgh School of Medicine, Director of the UPMC Eye Center, and the Eye & Ear Foundation Endowed Chair of Ophthalmology. He is known worldwide for his expertise in vision restoration techniques and has developed several interventions.
While the field is in a stage where we are starting to treat patients with gene therapy, unfortunately not every patient with a genetic disease can access it. A goal of gene therapy is to correct a gene defect, using a vector, which is a modified nonpathogenic virus that contains the code for protein. It is packaged and gets inside the cell, where the protein is expressed and used to correct retinal degeneration (RD). There are many ways to do this.
Vectors are delivered in different ways to the eye: subretinal, intravitreal, and suprachoridal. The current most classical way is to inject under the retina, something that has been done for many other conditions for decades. Another way is to inject directly into the vitreous cavity of the eye. A new approach that has been developed over the past few years is suprachoroidal, where the vector is injected close to the choroid, the vascular nurturing tissue under the retina.
Each approach has its advantages and disadvantages. Some are a bit more invasive, and some lead to more inflammatory responses, while they also have safety advantages.
“In age-related macular degeneration, the main goal of people using gene therapy is not to correct the cause of the disease – because we don’t actually know precisely the cause – but to treat the complications of a disease, namely the atrophy of a retina or proliferation of new vessels,” Dr. Sahel said. The goal is also to limit the number of injections.
Pittsburgh has several existing gene therapy trials. Jay Chhablani is overseeing one of them.
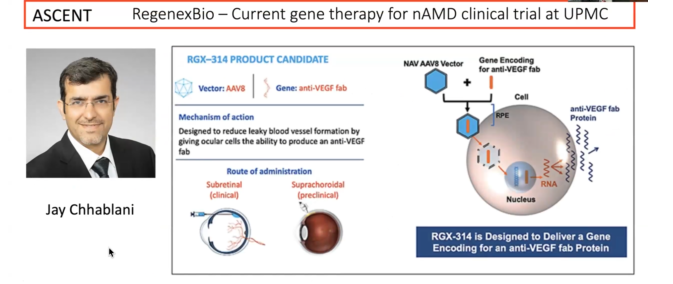
“The main focus for many of us is on genetic diseases of the retina,” Dr. Sahel said. “We are trying to target various stages of the disease.”
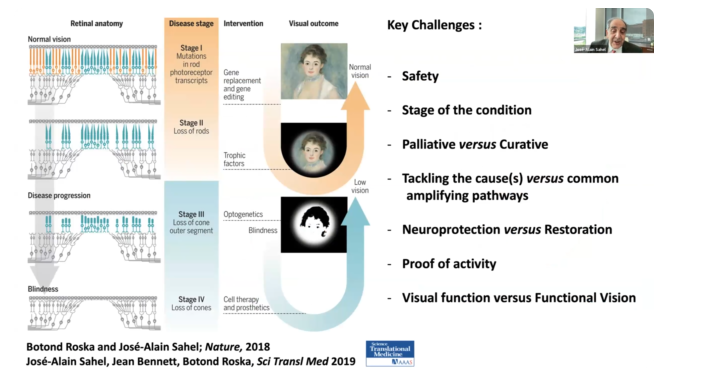
These cells are affected by mutation, which leads to the loss of dark-adapted vision (the way we see at night). Over the course of time, peripheral vision is lost and eventually color vision is too. The central vision field becomes narrower over time and ultimately can lead to a total loss of vision.
The ideal therapy is to correct the gene defect. If this is done early enough, the course of disease can be stopped, and the patient can potentially recover or keep useful vision. This is gene replacement, which is adding the gene that is missing or gene editing, which is is trying to correct the typo in the genetic code that is leading to the disease.

One of the key projects of Dr. Sahel’s team is to protect the remaining cells. He has been working on this for years.
Gene Therapy for IRD
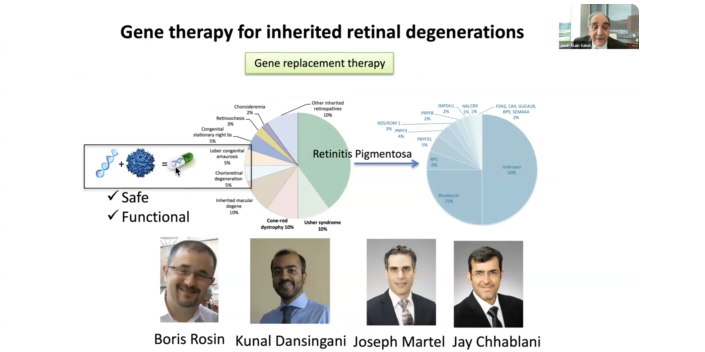
Currently only a small number of patients can be treated with existing approaches. The team focuses on the main one, which is early onset disease that occurs in children leading to early on certain loss of vision. Luxturna was approved in the U.S. in 2017 and in Europe in 2018. It has demonstrated a significant on vision, especially in dark-adapted vision. “This has been one of the first gene therapies approved across the world, with the U.S. being the first country,” Dr. Sahel said. Pittsburgh has started to treat patients with Luxturna. Our team, led by Michelle Alabek, and a patient navigator, helps ensure that every patient has access to a therapy.
This is still a small number of patients, with many who do not have access to gene therapy because it is not yet validated for their specific gene defect. There are so many gene defects. With retinitis pigmentosa (RP) alone, there are 70 different genes, with different gene defects in each.
Practical Challenges to Gene Therapy for IRD
Indeed, there is a complexity and large number of mutations. There are still a lot of unknown genes. About 15-30% are yet to be identified. The stage of intervention is also important because there is a state where it is too late to intervene for the patient to benefit.
Dr. Sahel’s team has been concerned about possibly having to treat patients later in the course of the disease. As a result, they have been working for years on trying to protect the central vision. They identified a key mechanism that is able to maintain viable photoreceptors. This therapy is now in clinical trials.
Dr. Joseph Martel is principal investigator on the study that is trying to rescue the chorids by providing the signaling that is underlying the survival. This is in clinical trials, with key controls in Paris and Pittsburgh. A few other centers are opening in the U.S. like Miami in the coming days.
Optogenetics and Artificial Vision
Another approach is optogenetics, “where we express in the remaining cells of the retina proteins that are sensitive to light,” Dr. Sahel said. GS030 optogenetic therapy is combined gene therapy and a medical device to restore retinal light sensitivity.
PIONEER: First in-human clinical trial of GS030 optogenetic therapy
- Phase I/IIa, open label, unmasked, non-randomized, dose-escalation study
- 3 sites: UK (Moorfields Eye Hospital, London), France (Hopital des XV-XX, Paris) USA (University of Pittsburgh Medical Center)
- Study population: end-stage non-syndromic RP
- Single intravitreal injection in the worst-affected eye
- Primary endpoint: Safety and Tolerability at Year 1
- Secondary endpoints: visual function, orientation and mobility, OCT, quality of life, immune response
Dr. Martel was PI in the study that demonstrated in the first patient, treated in Paris, the ability to move from being totally blind to being able to see. Written up in Nature Medicine, a blind RP patient had partial recovery of visual function after optogenetic treatment. The 58-year-old was diagnosed with RP 40 years ago and had light perception when enrolled in the study. After GS030 optogenetic therapy, he had partially repaired vision. He was able to locate and count objects on a table and identify crosswalks in the street. “This is a key milestone on the path to restore vision,” Dr. Sahel said. Other patients who were previously blind have experienced a similar benefit.
They were able to show that this is correlated with reactivation of the brain in the visual area, thanks to the support of leading cognitive neuroscience in Paris and Pittsburgh (Dr. Marlene Behrmann). Now data is being collected and the therapy is being improved to provide much better vision. Dr. Sahel called this very promising and said they are working on the next generation of this promising therapy.
Gene Therapy for PRPF31 Retinitis Pigmentosa
The last part of the presentation was by Leah Byrne, PhD, Assistant Professor in the Department of Ophthalmology, with secondary appointments in the Departments of Neurobiology and Bioengineering. The Byrne Lab develops gene-based approaches, including viral vector-mediated gene delivery and genome editing, to interrogate the biology underlying retinal disease and treat inherited blindness.
A new tool has been developed where instead of replacing a gene, the mutation can be rewritten. Dr. Byrne prefaced the next part of her talk by clarifying that the virus referred to in these experiments is called adeno-associated virus (AAV), something we live in harmony with most of the time. AAV characteristics:
- Replication incompetent (it cannot make more copies of itself)
- Nonpathogenic (does not cause disease)
- Infects retinal neurons
- Permanent expression but non-integrating
All this makes AAV an excellent option for retinal therapy. “The promise of gene therapy is that you’ll get a permanent solution to the problem after a single injection,” Dr. Byrne said. “In theory, if everything works as we hope it will, then you will deliver the virus one time. Then that therapeutic gene is going to stick around in the retinal cells the rest of the life of the patient. I think gene therapy holds a lot of promise for that reason.”
PRPF31 RP (RP11)
“It’s a rare disease but it’s one of the most common rare retinal diseases,” Dr. Byrne said of PRPF31 RP.
In terms of its genetics:
- Caused by mutations in PRPF31, a splicing factor
- Over 100 causative mutations in PRPF31 have been identified
- Prevalence: PRPF31 mutations account for 2.5-10% of autosomal dominant RP
- Current management: Monitoring of disease progression
Since there are not a lot of treatments for this disease, it represents a major unmet need in the medical field. “I think gene therapy holds a lot of promise for treating this disease.”
RP11 is caused by haploinsufficiency. As Dr. Byrne explained, everyone carries two copies of the PRPF31 gene. If you have a mutation or just one copy of that gene, then you can either have the disease or be normal. Whether or not you have the disease is determined by if that second copy is a high expressing copy or a low expressing copy. So, if you inherit from your parents one mutated copy and you have a low expressing copy on the other allele, then you are going to have disease. That’s called haploinsufficiency. “That also means that this is sort of a naturally occurring model of how gene therapy could work, because if you inherit one mutated gene and the second copy is high expressing, you do not have disease,” Dr. Byrne said.
In other words, if they are able to increase the amount of PRPF31 by a little bit, they are very likely able to treat this disorder.
One of the biggest problems in the field is the lack of an animal model to test these gene therapies. Most of the time mice is used in research, but they cannot be used here. If mice have no copies of PRF31, they do not survive. The second copy of the gene tends to be high expressing in mice, so if they only get one mutated copy of the gene, they either do not have the disease or only get a mild form of it. Testing the gene therapy is an important step for preclinical investigation, so Dr. Byrne’s team set out to create a mouse model using CRISPR.
Using CRISPR/Cas9 to Make a Mouse Model of PRPF31
CRISPR is basically a pair of molecular scissors used to cut the genome. The team put CRISPR into mice and cut out a little piece of the PRPF31 gene. This inactivates the gene and leads to disease in the mice. A gene therapy using a virus was then delivered.
This was done with a bunch of different mice successfully. But it is important to test this in more than one animal, particularly a large one, before moving into the clinic. Mice are so different from people, but primates have eyes that are very similar to humans. Dr. Byrne’s group is using the same approach in primates. If they are able to create a primate model and show rescue, this will be stronger evidence that the therapy will work on people in clinic. “Evidence shows that we’re moving toward creating a monkey model that we can use to test these gene therapies even further and show even more evidence it’s going to work in patients,” Dr. Byrne said.
Conclusions and Future Directions
- CRISPR-Cas9-based approach can be used to create mouse models of PRPF31 that model the main features of RP11
- Gene augmentation rescues retinal structure and function in PRPF31-KO mice
- The same approach can be used in primates. Can rescue be achieved in primates?
- Can this therapy be translated into the clinic?
“We always have an eye on the clinic and are hoping to move this into the clinic and towards patients as quickly as possible,” Dr. Byrne concluded.