
A lot of progress has been made in the past few years on retinal degeneration, Dr. José-Alain Sahel said at the beginning of the Eye & Ear Foundation’s May 18th webinar titled “Retinal Dystrophies: Introduction, Therapies and Ongoing Research.”
Hereditary Retinal Dystrophies
Boris Rosin, MD, PhD, Clinical Instructor of Ophthalmology, part of the Retina and Vitreous Service in the UPMC Eye Center, explained hereditary retinal dystrophies and visual function testing. He started by describing the functional cells of the retina. The photoreceptors are divided into cone cells (responsible for sharp color vision) and rod cells (responsible for night vision), which deal with phototransduction. “That is basically taking the light stimulus picture that we see and translating it into a language that the brain understands, which is that of electrical potentials,” Dr. Rosin said.
The process of phototransduction, comprised of multiple protein cascades, illustrates how much can go wrong. A mutation in any of the participants in this and other processes can and indeed does cause an inherited dystrophy, Dr. Rosin said.
The basic building block of phototransduction is the opsin, a protein capable of reacting to light. The different types of opsins determine the spectral properties of the photoreceptor expressing it, namely to which wavelength of light will it react most strongly.
Visual Function Testing
Visual function testing is a wide field comprised of many tests. The most common one is the Electroretinogram (ERG), put forth by a British scientist named Ragnar Granit in the 1930s (He received the Nobel prize in Medicine and Physiology for his efforts in 1967). ERG is performed by applying recording electrodes on the cornea, in fornix, on the canthus, and even the skin. A ground electrode is also usually on the ear. The retina is then stimulated by different types of light, allowing separation of the rod and cone photoreceptors under different stimulus conditions.
Retinitis Pigmentosa Epidemiology
Retinitis pigmentosa (RP) is the leading cause of inherited blindness. The incidence of RP is about 1/5000 live births, not including syndromic types (Usher syndrome, Leber’s congenital amaurosis, and others, which include RP and an additional ailment, e.g. Usher syndrome – RP with congenital hearing difficulties of various severity). The incidence of RP together with the syndromic subtypes is about 1/4000 with syndromic types. The total incidence of hereditary retinal dystrophies is about 1/3000. RP is mainly autosomal recessive (AR) inherited, however autosomal dominant (AD), which are usually milder, and X-linked, usually more severe, variants exist.
What happens in patients with advanced RP? The three cardinal signs of RP on physical examination are waxy pallor of the optic nerve, attenuation (thinning and uneven diameter), and deposition of pigment in what is called “bone spicule pattern” due to its resemblance to bone tissue in histological preparations.
Useful Tests
- Optical coherence topography (OCT) – Utilizes light waves in a manner similar to ultrasound utilization of light waves, providing a high-resolution image of the retina. When looking at OCT images of RP patients of different ages, providers can see the retina becoming more atrophied with age.
- Electrophysiology – The gold standard for diagnosis. The ERG in advanced RP is extinct, showing no response to any type of stimulus.
- Visual field estimation – A mapping of the patient’s visual field through stimulation by light presented at different locations.
Achromatopsia
Achromatopsia is a rare (about 1:30,000-40,000) retinal disease of cone dysfunction characterized by:
- Severely reduced visual acuity
- Photophobia – extreme sensitivity to light
- Nystagmus – uncontrolled rhythmic eye movements
- Markedly impaired color vision
There is a much higher incidence in Israel (about 1:5,000) due to the two religious populations (Chassidic Ashkenazi Jews and Muslim Arabs), which tend to intermarry. The largest known incidence is Pingelap Island (1:10). Oliver Sacks, a famous neurologist and author, wrote a book called The Island of the Colorblind, which talks about this fascinating community.
Therapies and models of Hereditary Retinal Dystrophies
The retina is an ideal target for gene therapy, because it is a small, confined tissue that is accessible by injection. Clinical and functional follow up is also readily available. In simple terms, gene therapy involves taking a virus, emptying it of its infective component and loading it up with a copy of a gene to be expressed in the target tissue (in this case, the retina). The target tissue is then infected with the virus and starts expressing the gene.
Gene therapy could potentially be used for:
- Gene replacement – replace a mutated protein with a normal one
- Delivering neurotrophic/survival factors – such factors can prolong the life of photoreceptors
- Reducing expression of mutated proteins by introducing gene expression control proteins
- Delivering photosensitive proteins to retinal cells previously not expressing such proteins
Dr. Rosin shared some gene therapy studies, including ones with canines and sheep. Dr. Sahel’s optogenetics study falls in this category as well.
Stem cells are the ultimate replacement therapy, with human embryonic stem cells (hESCs) a platform for treating retinal disease. The objective is to develop technology that will facilitate the use of hESCs for cell therapy. There are three main target cell types of interest:
- Photoreceptors (RP, AMD)
- Retinal pigment epithelium (AMD, RP)
- Ganglion cells (glaucoma)
Embryos are not needed these days for two main reasons: human embryonic stem cells are mainly derived in the lab. They have an unlimited amount of division potential. Cells can also be taken from a specific patient and induced to revert their cell fate, turning them into stem cells. Cell therapy is lagging behind gene therapy in the treatment of retinal dystrophies due to difficulties in proper integration of newly formed photoreceptors in the pre-existing neuronal circuit. Nevertheless, advances have been made in cell therapy for the replacement of simpler tissues, such as the use of cell therapy for the treatment of dry AMD, where it has shown promise in limiting the extent of atrophy in early-stage clinical trial.
A very hot field is retinal organoids, eye-like structures developing from stem cells in 3D culture media that have staggering applications. Retinal prostheses – both biological and artificial – are also being explored.
A new development in the field of microbiology is CRISPR, which is piggybacking on a mechanism employed by bacteria to correct gene defects caused by viral infections. Dr. Leah Byrne in the Department of Ophthalmology at the University of Pittsburgh employs these and other methods in many ways. Possible uses include:
- Correction of genetic defects
- Creation of new organoid models
- Creation of new animal models – Up until now, researchers were mainly reliant on very few naturally occurring models. However, CRSIPR could provide a potentially unlimited variety of models by means of introducing the mutation in question into stem cells. Such cells can then be used to create either new retinal organoids expressing such mutations or a whole animal expressing the mutation of choice. This will allow the research of diseases which previously had no good animal models. These make up the majority of the retinal dystrophies.
Clinical Trials
The second part of the presentation focused on clinical trials. Michelle Alabek, MS, certified genetic counselor, and coordinator of the adult retinal dystrophy clinic started with an overview of general terminology.
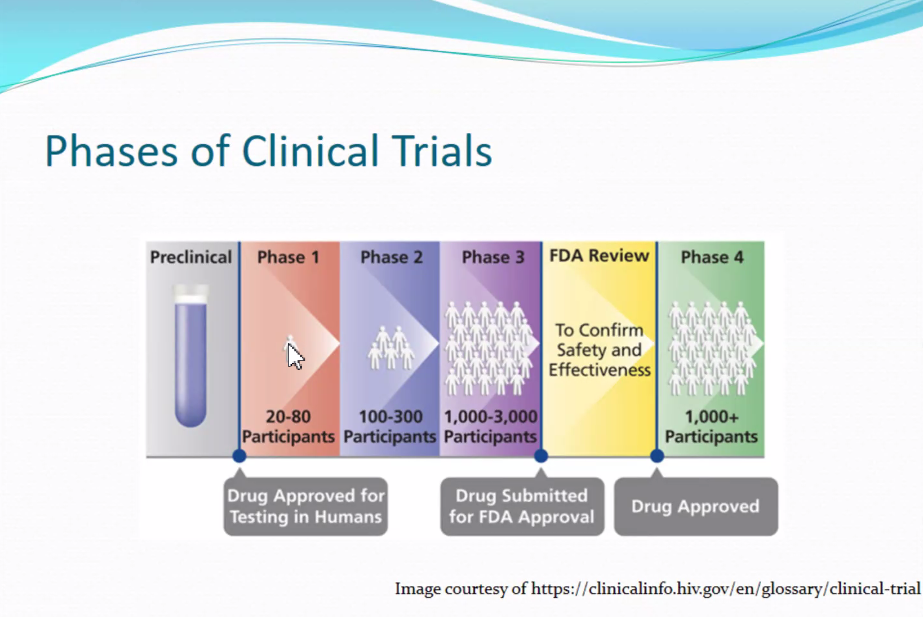
- Natural history study – Collects data in a very standardized way to understand and document the normal progression of the disease. Can be very helpful in laying the groundwork for future clinical trials.
- Clinical trial – When a human subject is given some type of intervention or treatment with the goal of evaluating the effects of that treatment.
- Protocol – The specific design of the study planned with the patient. Safeguards are in place with a very specific research question and how it will be answered or addressed.
- Informed consent – A document that explains to the potential subject or potential participant all the different risks, potential benefits, etc. that they may want to consider before deciding whether to participate
- Phases – Clinical trials done in various phases, progressing from one to the next. Usually, each successive phase has its own protocol.
- Inclusion criteria – Inclusion correctly refers to all the factors or variables that a patient or participant must meet to be considered eligible. In retinal dystrophy trials, a lot of the inclusion criteria are common things like age, diagnosis, genetic result, and visual acuity. There is a very specific range. Each protocol has its own inclusion criteria.
- Exclusion criteria – Individual factors that if a participant meets, would deem them ineligible for the trial. These are often longer or more comprehensive than inclusion criteria. They include:
- Other ocular disease
- Systemic disease
- Medications
- Pregnant or breastfeeding
- Other genetic diagnosis
- Prior treatment with investigational drug
- Past retinal surgery
- Actively participating in another clinical trial
Clinical Impacts of Genetic Testing
Once a genetic cause is identified, this impacts the diagnosis, prognosis, personalized management, treatment options, and risk for relatives. The more trials that are available, the more therapies that are moving forward and progressing, which is becoming increasingly relevant. “Genetic testing is really having more of an impact on deciding and making decisions about options that may be available to patients for new treatments,” Alabek said.
The Foundation Fighting Blindness website highlights all the active trials for retinal dystrophies.
Is a Clinical Trial Right for You?
To assess whether a trial is right for you, talk to your doctor to understand:
- Inclusion exclusion criteria
- Phase – Are you comfortable with this?
- Timing of intervention – For progressive conditions, there may be therapies less or more appropriate depending on your stage of disease. This means really understanding what type of intervention or therapy is being provided in the trial, and kind of comparing that and cross-referencing that to where you are at in the stage of your disease.
- Details of the trial – Weigh all the information, such as:
- Long term benefits vs short term risks
- Responsible provider
- Short term benefits vs long term risks
- Travel reimbursement
- Duration
An example of a Clinical Trial Timeline
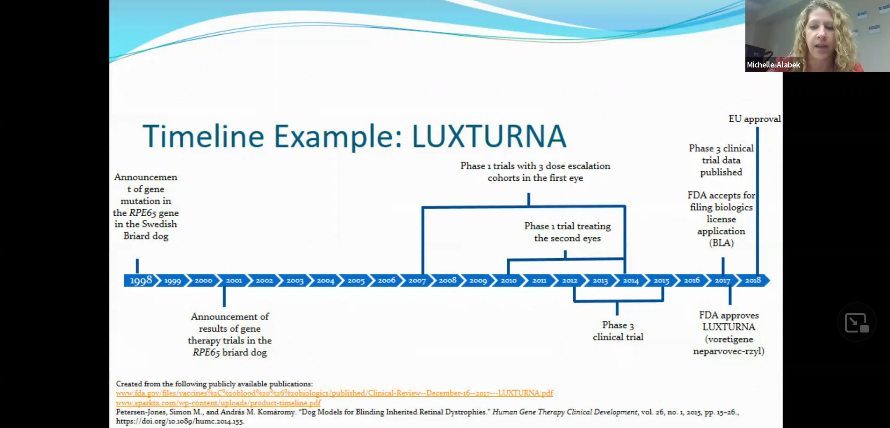
“Everyone, providers and patients alike, would prefer that this timeline is shorter,” Alabek said. Things should not be rushed in a way that jeopardizes patients’ safety. The process is done with a lot of intention and responsibility. There are some efforts, however, to try and expedite some of the progress made in this area. The Monaciano Consortium Symposiums, an international group of clinicians, scientists, and individuals that manage patients with retinal dystrophies are identifying key steps needed to advance treatment.
Another organization working on this kind of collaboration is the Foundation Fighting Blindness, which has the Foundation Fighting Blindness Consortium. Forty participating centers across the U.S. and internationally are working to conduct clinical trials collaboratively to accelerate treatment. Several clinical trials are open, and several others will start later this year.
Tips for Patients Interested in Clinical Trials
- Attend routine appointments with your ophthalmologist
- Gather as much information as you can about your diagnosis
- Stay up to date on advances in the field
- Do not feel like you have to jump at the first opportunity that is available
- If you are not eligible, remain hopeful!
Resources for Staying Up to Date
How UPMC Identifies Patients for Studies
- Review protocol
- Assess available data for patients
- Contact patient to discuss potential trial eligibility
- Send patient consent form to review
- Schedule conversation with physician
- Schedule screening visit
UPMC Retinal Dystrophy Clinic and Services

