At the start of the Eye & Ear Foundation’s August 6 webinar, “Inherited Retinal Conditions: Current Treatments and Future Research,” Dr. José-Alain Sahel, MD, Distinguished Professor and Chairman of the Department of Ophthalmology, said, “As this is not the first webinar we’re giving on this topic, we’re trying to update you on the recent progress.” Dr. Boris Rosin, MD, PhD, physician-scientist, introduced hereditary retinal dystrophies and visual function testing, followed by Morgan Brzozowski, MS, LCGC, genetic counselor and coordinator for the Retinal Dystrophy Consultative Services at the UPMC Vision Institute. Dr. Sahel then presented promising research.
Introduction to Hereditary Retinal Dystrophies
Our vision is basically governed by certain cells within the retina called the photoreceptors, or cells able to perceive light. We have three types: red, green, and blue. They are responsible for daytime and extremely sharp vision.
Photoreceptors within the retina are different. There are cone cells vs rod cells. The ability of photoreceptors to sense light comes from opsins, which are derived from vitamin A. When hit by light, photons change configuration and initiate a cascade that results in activating the photoreceptor. The process of phototransduction is how light is converted into electricity, the language of the brain. There are many different types of opsins, all characterized by a wavelength of light. Rod opsins are within the rod, while cone opsins are within the cone.
Many components of this cycle can be damaged, mutated, or impaired in genetic retinal dystrophies.
Electroretinography (ERG)
To diagnose these disorders, the retina’s function is tested, which translates the image seen into the language of the brain, which is light. This is done with electroretinography, which records the electrical activity of the retina in response to stimulation by light. It can discern between the rod and cone photoreceptors. Recording electrodes can be placed on the cornea, in fornix, canthus, and even on skin. Ground electrode is usually on the ear.
Stimulus conditions for rod ERG:
- Total darkness!!
- Dark adaptation 20-45 minutes
- Dim flashes, below cone threshold
- Preferably blue (~ 500nm) flash color
Stimulus conditions for cone ERG:
- Background lighting to suppress rods
- If after dark, allow light adaptation 10-20 min
- Bright flashes, above cone threshold
- White or red flash color
- Can use rapid flicker (usually >30Hz)
Case Studies
Dr. Rosin shared a case example that is “typical” of what he sees in the clinic. A 36-year-old male who is generally healthy complained of decreased visual acuity that was progressive and had only partial improvement with glasses. He also complained of stumbling and having difficulty going down the stairs. He had trouble playing ball games as a child. At night, he had severe disability, especially when in really dark environments (like camping). He had two brothers with a similar problem.
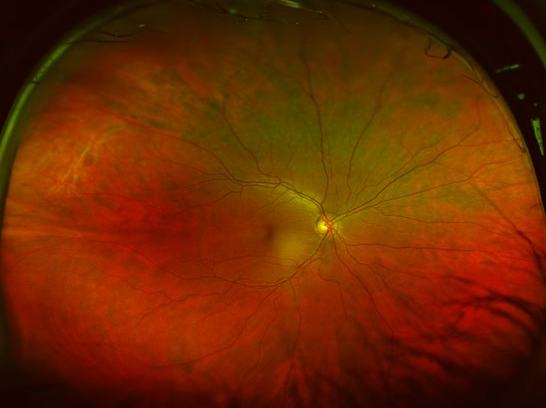
This patient’s visual acuity (VA) was 20/40. Anterior segment examination was performed. Fundus (retinal) examination revealed widespread retinal degeneration, optic disc pallor, and bone spicule-like pigmentation. Genetic testing found the FAM161A mutation, confirming the diagnosis of retinitis pigmentosa (RP).
RP Epidemiology
RP is the leading cause of inherited blindness. Its incidence is ~1/5000 live births, not including syndromic types (Usher syndrome, Leber’s congenital amaurosis, etc), and is ~ 1/4000 with syndromic types. Total incidence of hereditary retinal dystrophies is ~ 1/3000. RP is mainly AR inherited; however, AD (milder) and X-linked (more severe) variants exist.
“These diseases are exceedingly rare,” Dr. Rosin emphasized. Every specific mutation is less common.
Other Diagnostic Tools
Optical Coherence Tomography is a retinal scan that shows the layers of the retina. Electrophysiology is the gold standard for diagnosis. Visual field estimation is another diagnostic tool, used to look at visual fields – basically the field of vision where the person sees what is going on. Visual fields become progressively attenuated as the disease continues.
Case Study #2
Dr. Rosin pointed out that while he picked two case examples he felt were interesting, there are hundreds of different disorders that are very difficult to diagnose. He is faced with a diagnostic challenge daily.
In this case study, a 27-year-old male college student was diagnosed with “retinal degeneration” at the age of nine months when his parents noticed him squinting in the sun. He complained of severe photophobia, decreased visual acuity, poor color vision, and had nystagmus since early childhood. He was usually healthy, with no known drug allergies, and did not take any medications or smoke.
Upon a physical examination and ancillary testing, his VA was 6/60, confirming the diagnosis of achromatopsia. This very rare condition (~1:30,000-40,000) is a retinal disease of cone dysfunction characterized by severely reduced visual acuity, photophobia, nystagmus, and markedly impaired color vision. In an ERG, the full-field response is composed exclusively of rod response with the cone response being practically absent.
The largest known incidence of this disorder is the Pingelap Island (1:10). The famous neurologist Oliver Sacks – who wrote a lot of books about medicine – wrote a book about this condition and talked extensively about the island in his book, The Island of the Colorblind. “It’s a very cruel twist of fate,” Dr. Rosin said, “because [the people who live] in this amazing, beautiful Pacific island likely see it completely blurred in shades of gray and white.”
Genetic Testing in Inherited Retinal Dystrophies (IRDs)
Upwards of 300 genes are implicated in many IRDs, both syndromic and non-syndromic, with many different inheritance patterns. There are many overlaps because these genes can cause multiple conditions depending on where the exact genetic change occurs.
Because of this overlap, genetic counselor Morgan Brzozowski recommends taking a panel-based approach to genetic testing as opposed to targeting a single gene or doing broader scope testing. This method allows for analysis of many genes that can cause similar symptoms across various diagnoses and is often the first line approach to genetic testing.
Panel-based testing curates sets of genes known to be associated with certain conditions or a collection of clinical symptoms. Panels are limited to the genes known to cause specific conditions. Many labs have IRD panels, but no two panels at these labs are the same. The genes are constantly being added and removed at any given time.
When thinking about panel selection, counselors like Brzozowski first think about the genes included on the panel. Most genetic testing reports will include the numbers of genes that were tested, along with a list of what they were. Another thing they look at when deciding which test to order is the testing technology, as sequencing technology differs between labs. Some regions of genes to sequence can be very tricky, and additional variants are hard to sequence in certain genes as well.
Counselors also consider the quality of the specific genetic testing report. Labs have different access to resources they utilize when interpreting genetic testing results and writing the reports that are ultimately shared with care providers. Certain labs have more experience with genetic testing for IRDs. Having knowledge of and access to one of those labs may result in a higher quality report.
The cost of genetic testing can be very expensive, so this is taken into consideration as well as the financial assistance plan through that specific lab. Access to genetic testing for IRDs can be obtained through insurance, self-pay, or sponsored testing. If through insurance, a prior authorization process is initiated, whereby the counselor provides the insurance company with clinical information and family history. They have pretty good success in getting the authorization, but just because testing is authorized does not mean there is not a cost associated with that testing.
Self-pay means paying the lab directly, but the fee can be different depending on the panel and the lab.
Two sponsored testing programs are available for IRDs. They have different eligibility requirements, with limitations and benefits to each. Both involve data sharing, helping improve access to genetic testing.
One program is My Retina Tracker Registry, through the Foundation Fighting Blindness. Patients need a clinical diagnosis of an IRD listed as part of the program and to not have undergone genetic testing with a panel consisting of 32 or more IRD genes within the last three years.
The other program is Invitae unlock, which requires individuals to have a diagnosis of IRD or a family member with a diagnosis. This program is dependent on insurance coverage. If a clause in the patient’s insurance states that genetic testing is not covered, then this program may be an option. A genetic counselor can provide further information on eligibility criteria for both programs.
Genetic Testing Results
Three different results can come back, and it is “really important for us to interpret those results based on the clinical context that we have from the individual’s history,” Brzozowski said.
A positive result, meaning genetic variant(s) were identified and explain the individual’s history, occurs 70% of the time. Inconclusive is when the identified gene variant(s) could explain the individual’s history. Testing on family members is often recommended in this case. Or the result could be negative, with no identified gene variant(s) to explain the individual’s history. A negative genetic testing result does not mean there is not an underlying genetic cause, just that they were not able to identify the cause using current testing.
When a genetic cause is identified, it is not a crystal ball that will provide all the information but is a roadmap with guidance on what the path might look like moving forward. It helps with diagnosis, prognosis, risk for relatives, systemic manifestations, and treatment options.
With systemic manifestations, IRDs can be isolated to just the eye, or they can be syndromic, meaning they affect other parts of the body. Some systemic manifestations that result in proactive screening and management recommendations involve cardiology, audiology, and some other specialties depending on what the exact gene is. “It gives us more information on symptoms expected in the future and allows us to provide better surveillance for such symptoms,” Brzozowski said.
Treatment options include some targeted gene therapies that are available, whether that means FDA-approved or clinical trials. In some cases, there are surgical implications.
However, the challenges of genetic testing are that not all IRD genes are known, not all patients with an IRD will have a positive result, and not all genes will have a targeted trial.
“Over time, we are getting better at understanding genetics in ophthalmology and IRDs,” Brzozowski said. As knowledge continues to develop, there are more opportunities. Consider updated genetic testing because of identification of new genes, advancements in sequencing technology, development of new therapies, and access to sponsored testing.

When considering doing updated genetic testing, it can be very helpful to sit down and talk to a genetic counselor. “At UPMC, we are very lucky to have a team of four in ophthalmology, thanks to the excellent support we have from physicians here at the Vision Institute and Children’s,” Brzozowski said.
Current Treatments and Future Research in Retinal Degeneration
“As a reminder, vision in humans is both central and very full vision,” Dr. Sahel said. Central vision is very high acuity and is where we read and recognize faces. We also rely on peripheral vision for a lot of activities. The retina allows us to see in the dark and with strong lighting. All of this relies on the distribution of photoreceptors. Many conditions affect the central retina, called macular dystrophies.
AMD – where mostly the central retina is affected – is seen in 30 million people, with 10-20% severely impaired. Around 1.5 million people have retinitis pigmentosa and are legally blind by the age of 40.
The ideal therapy would be to correct the gene defect by utilizing mostly gene therapy to bring back the normal gene or correct the mistake in the genetic code. If you do this early enough and are able to target as many cells as possible, it could be considered a cure. One product has been able to address this quite well – Luxturna for RPA65. UPMC is a center that provides it. This is only for a very small number of patients. Most of the time, it mostly restores some visual function.
Many genes are currently subject to trials, and the Department of Ophthalmology is involved in many of them. They are eagerly awaiting results, hoping some will turn out to be positive.
One stage of disease where it is likely too late to rescue the photoreceptors because they are already advanced in terms of degeneration has fueled Dr. Sahel’s work for many years – protecting the remaining cells. This is called neuroprotection using trophic factors. Prosthetic vision, cell therapy, and optogenetics have all been tried. There are many challenges, however:
- Safety
- Stage of the condition
- Palliative vs curative
- Tackling the cause(s) vs common amplifying pathways
- Neuroprotection vs restoration
- Proof of activity
- Visual function vs functional vision
The retina is very fragile. Everyone wants to get the treatment but should not put themselves at risk, Dr. Sahel said. He advised asking a lot about the risks and benefits.
Gene Therapy for IRDs
Gene therapy is trying to address the gene defect. There have been some successes, but there are practical challenges:
- Complexity and large number of mutations
- Unknown mutations
- Timing of intervention
- Demonstration of unequivocal benefit
Retinitis Pigmentosa is a rod-cone disease. Most mutations are rod specific. Patients initially lose peripheral and duct-adapted vision. They are not able to see in the dark. When they lose some peripheral vision, they have tunnel vision, where they can see in the middle pretty well. Over time, this channel becomes more and more narrow and could eventually lead to severe loss of vision. Dr. Sahel has been working for many years on trying to protect what is left and looking at what happens in the course of a disease.
When it comes to the potential of cone-directed therapeutic strategies, Dr. Sahel shared quotes by Paul Sieving, “50% cone loss compatible with an acuity of 20/20” and “95% cone loss compatible with a correct orientation and discrimination performance.” Alan F. Wright, in a Nature Genetics 1997 article said, “Preserving cones would prevent 1.5 million people worldwide from becoming blind, since in an age of artificial lighting, we function very well without rods.”
Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Dr. Sahel’s team in Paris and now in Pittsburgh has been working on trying to understand why the cells that are not affected by the gene defect are degenerating. They identified that there is a signal made in the normal retina when the rods are still alive. This led to a therapy where they try to restore the signaling to bring back the signal that was lost. “My group believes the goal is to restore the part of the photoreceptors that respond to light,” Dr. Sahel said. This led to a clinical trial that is ongoing in Paris, Pittsburgh, and a few places across the world. They are currently in the middle stage. He called the trial safe and hopefully promising.
Beyond neuroprotection, another stage where remaining vision is no longer able to be protected because cone photoreceptors have already undergone significant degeneration is restoring vision through optogenetics or trying to reactivate the remaining cells. This is done by using a “very peculiar” mechanism that exists in very rudimentary organisms like algae where they are able to detect the light and move away or toward the light.
There are four different kinds of optogenetic therapy: cones, bipolar cells, all amacrine cells, and ganglion cells. Ganglion cells – which connect the eye to the brain – do not normally respond to light, but even in the advanced stages of a disease, they are still alive and express proteins responsive to light. Therefore, treatment using ganglion cells and optogenetics means using light gated channels to transform RGCs into depolarizing photoreceptors. In RP patients, retinal neurons survive years after photoreceptor degeneration.
GS030 optogenetic therapy is combined gene therapy and a medical device to restore retinal light sensitivity. They showed there is a signature in the retina. After 10 years of work, they started a clinical trial. The mechanism of action of GS030 combined therapy is as follows:
- Intravitreal injection of viral vector and RGC transfection
- Expression of light-sensitive ChrimsonR on RGC surface
- Induction of neuronal signal by photostimulation of bioengineered RGCs
It basically restores light sensitivity of the retina by modifying and training RGCs to act as photoreceptors. A 58-year-old blind patient diagnosed with RP 40 years ago who had light perception when they enrolled in the study partially regained vision after this therapy. They could locate and count objects on a table and identify crosswalks in the street.
Many groups are now working on the same approach. Some are developing products. There is a big effort in the Department to increase the specificity and resolution of images and get to the next generation of optogenetics.
Artificial Retina
Another approach is the artificial retina, or retinal implants with a retinal prosthesis. Implants stimulate the enormous cells in the retina. Results have been successful but not with very high resolution.
Another implant was developed with Stanford in which a chip is implanted with a photodiode that responds to light. PRIMA clinical trials in Europe and the US – including Pittsburgh – showed improvement of vision that was significant and impressive and should let patients retain their peripheral vision. It was done in patients with central macular involvement in AMD. A multicenter trial in Europe with 38 patients just completed, with improvement of visual acuity in the majority of patients.
In short, a lot of tools have been developed to document the ability to restore useful vision.
“We are trying to develop approaches that are meaningful for the patient that are going to make a difference in their daily lives,” Dr. Sahel concluded. “It is a very long roadmap that is not going to be easy. This is a lot of work with a lot of people. We benefit from the support of EEF and many donors, NIH, and other institutions, because a lot is needed to help regain or retain some of our vision.”